CUT&RUN
CUT&RUN stands for cleavage under targets and release using nuclease. The technique developed from the Henikoff Lab offers a new approach to pursue epigenetics. CUT&RUN introduces some major modifications to eliminate shortcomings inherent to ChIP-seq. It is simple to perform and inherently robust, with extremely low backgrounds requiring only ~1/10th the sequencing depth as ChIP. CUT&RUN is cost-effective for transcription factor and chromatin profiling.
You are new to CUT&RUN? Learn more about this application in the following sections or jump directly to our webinar series covering CUT&RUN and CUT&Tag.
CUT&RUN Antibodies
A range of antibodies validated for use in CUT&RUN assays.
CUT&RUN Sets
CUT&RUN made easy - with our sets & components
CUT&RUN ConA Beads
Magnetic ConA Beads for usage in CUT&RUN assays
CUT&RUN - Better than ChIP-seq!
CUT&RUN Workflow: Extraction Without Fragmentation
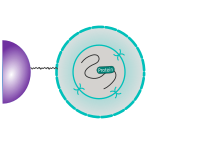
CUT&RUN is performed in situ on immobilized, intact cells without crosslinking. DNA fragmentation is achieved using micrococcal nuclease fused to Protein A and/or Protein G (pAG-MNase). The fusion protein is directed to the desired target through binding of the Protein A/G moiety to the Fc region of an antibody bound to the target. DNA under the target is subsequently cleaved and released and the pAG-MNase-antibody-chromatin complex is free to diffuse out of the cell. DNA cleavage products are extracted and then processed by next generation sequencing (NGS).
The standard CUT&RUN protocol is primarily intended for profiling of histone proteins and transcription factors that bind to chromatin. Reliable detection of rare transcription factors, transient interactions, or proteins in complexes that are not directly associated with DNA have proven difficult using CUT&RUN on uncrosslinked samples. Preserving protein-protein and DNA-protein interactions using crosslinkers on the other hand can introduce artifacts. The following depiction shows the standard CUT&RUN protocol:
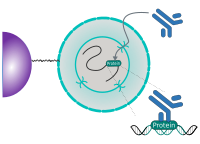
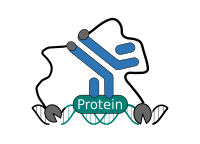
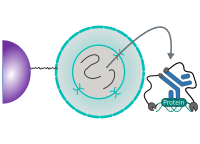
CUT&RUN Low Volume and Urea (CUT&RUN Lov-U) is a modification of the original procedure aimed at difficult to characterize interactions in situ while avoiding crosslinking. Initial nuclear extraction minimizes sequestration of the primary antibody and pAG-MNase by non-not chromatin-bound targets in the cytosol. Low volumes throughout the protocol facilitate parallel processing of samples which benefits scalability and reprodcibility. Lastly, urea is used as chaotropic reagent to denature samples in situ and release CUT&RUN products, followed by DNA clean-up on beads.
One of the drawbacks of CUT&RUN is the need to end-polish and ligate sequencing adapter prior to the preparation of a sequencing library. A combination of the CUT&RUN protocol and tagmentation by a hyperactive Tn5 transposase resulted in the CUT&Tag method. Cells are immobilized using magnetic Concanavalin A beads and reversibly permeabilized using digitonin. Instead of the directed nuclease cleavage however, DNA is fragmented by a pA/G-Tn5 loaded with sequencing adapter duplexes. Sequencing adapters are attached to the DNA fragments directly during tagmentation. No further DNA end processing is necessary and the fragments can be used for sequencing library preparation. In addition, CUT&Tag is inherently less sensitive to endogenous DNA damage than CUT&RUN because the transposition takes place on intact double stranded DNA molecules.
Learn more about CUT&Tag: Workflow, Advantages, Products
CUT&RUN Antibodies
A range of antibodies validated for use in CUT&RUN assays.
CUT&RUN Sets
CUT&RUN made easy - with our sets & components
CUT&RUN ConA Beads
Magnetic ConA Beads for usage in CUT&RUN assays
Discover our CUT&RUN Antibodies!
Show moreCUT&RUN and CUT&Tag Target Antigens
Histone modifications such as the repressive H3K27me3 or H3K4me3 associated with active promoters are commonly used antigens for both CUT&RUN and CUT&Tag. Additional antigens include transcription regulators like transcription factors CTCF or SOX2, chromatin modifiers like HDAC2, or proteins involved in nucleic acid metabolism such as Pol II. G-quadruplexes and m6A are among non-protein targets. Tags like e.g. FLAG, HA, or GFP are options for recombinantly expressed target proteins.
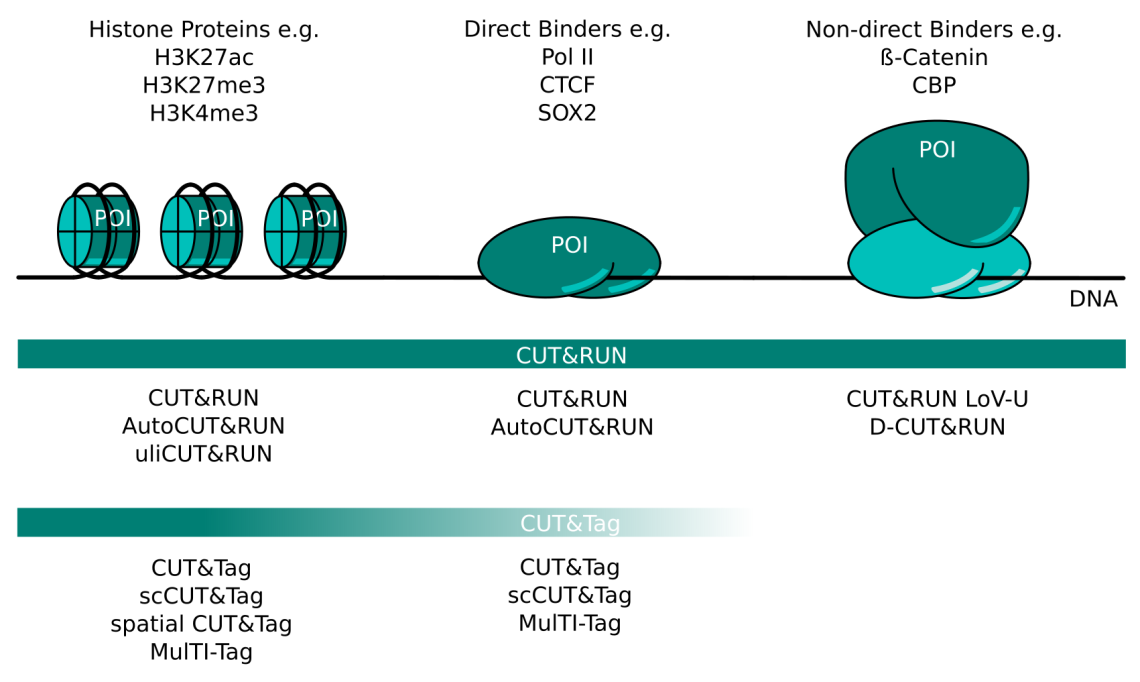
CUT&RUN and CUT&Tag Targets and Method Variants. Several variations of CUT&RUN and CUT&Tag have been described to better serve certain kinds of proteins of interest (POI). Histone proteins are very abundant and tightly bound in chromatin. Any CUT&RUN or CUT&Tag variant generally works well with these targets; some of the available methods are listed here. POIs that bind DNA directly like transcription factors or RNA polymerase II can be mapped well with CUT&RUN. The high salt concentration necessary during CUT&Tag wash steps can make this method more challenging for these targets. Non-direct binding POI like beta-catenin or CBP are the most challenging targets and necessitate CUT&RUN protocol adaptations like those listed here. CUT&Tag is less suited for non-direct binding POIs.
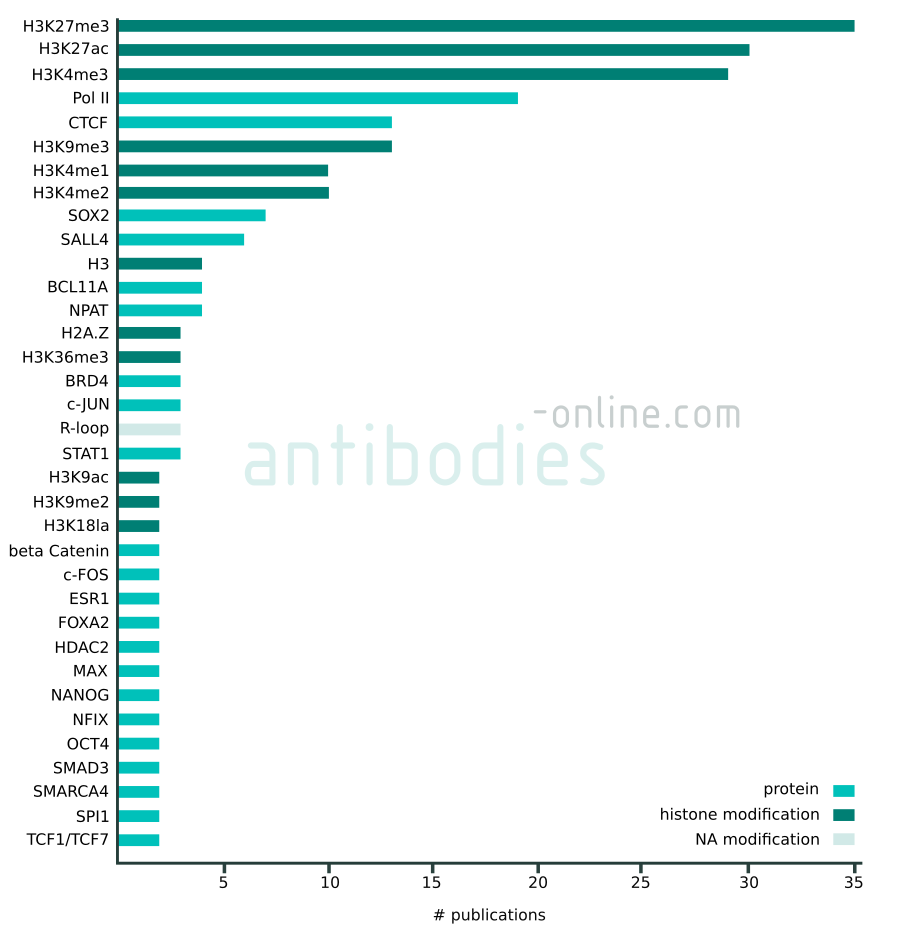
Targets that appear in at least two independent publications in context with CUT&RUN and CUT&Tag. The data represented in this figure is based on 137 freely accessible journal articles published from 2018 to 2022. Protein tag targets are not included.
Importance of Antibody Selection

Your desired target and application combination is not available yet? antibodies-online supports the validation of antibodies for CUT&RUN. Together with customers and manufacturers we are currently running more than 50 validation projects. And new ones are added almost weekly. You can participate in our independent validation initiative (IVI). Propose and perform a validation experiment (CUT&RUN) and receive full reimbursement for the validated antibody. Contact us via email without any obligation!
CUT&RUN: Frequently Asked Questions
- How do CUT&RUN and CUT&Tag work?
- What are the advantages and disadvantages of CUT&RUN and CUT&Tag compared to ChIP-seq? How do I choose between both methods?
- Can CUT&RUN and CUT&Tag replace ChIP-seq? Does CUT&RUN work for transcription factors, do-factors and other indirect binders?
- Why is the DNA yield so low?
- How do I choose a primary antibody for CUT&RUN or CUT&Tag?
- How can I validate that my primary antibody is working for CUT&RUN?
- Why do I need a negative control antibody? Why not just use a no-antibody control?
- Can I replace the antibody negative control for CUT&RUN using a knock-out (or knock-down) of my protein?
- Do I need to use a secondary antibody for CUT&RUN and CUT&Tag?
- One of my antibodies is mouse. Has your pAG-MNase good affinity for mouse antibodies, or do you advise to use a rabbit anti-mouse secondary antibody?
- Should I include heterologous spike-in DNA for quantitation?
- Is it possible to fix the cells prior to immobilization?
- Are there protocols to make this work for tissue, where I cannot necessarily isolate single cells?
- Instead of the proteinase K digestion can I denature the proteins in the CUT&RUN product complexes by heat?
- What is preferable for DNA extractions prior to library preparation: extraction using phenol-chloroform or affinity purification using a column?
- Is it possible to do single-end instead of paired-end sequencing of the CUT&RUN libraries?
Get our free CUT&RUN and CUT&Tag handbook now!
Show moreHow do CUT&RUN and CUT&Tag work?
CUT&RUN combines targeted fragmentation of genomic DNA in situ with high-throughput sequencing. Intact cells are immobilized on magnetic beads, e.g. through binding the lectin Concanavalin A, and subsequently permeabilized the cell membrane using a mild non-ionic detergent, typically digitonin. An antibody specific for the target of interest guides a protein A and/or protein G MNase fusion protein to the relevant sites for directed chromatin fragmentation. Once the MNase is bound via the antibody close to the intended DNA cleavage site, it is activated by the addition of divalent metal cations such as calcium. The digestion products diffuse out of the cells and analyzed using high-throughput sequencing.
CUT&Tag uses a protein A and/or protein G Tn5 transposase fusion protein instead of the MNase for chromatin fragmentation. The advantage is the omission of sequencing adapter ligation since they are part of the transposome itself and are attached during tagmentation. Sequencing and data processing are similar to CUT&RUN.
What are the advantages and disadvantages of CUT&RUN and CUT&Tag compared to ChIP-seq? How do I choose between both methods?
Advantages of CUT&RUN and CUT&Tag compared to ChIP-seq are a better signal-to-noise ratio, higher sensitivity, a wider dynamic range, a lower requirement of sequencing reads and cell number. Because it is possible to get meaningful data from fewer sequencing reads, CUT&RUN and CUT&Tag necessitate considerably less input material, which makes these methods preferable over ChIP-seq for rare samples.
CUT&Tag has the advantage that the sequencing primers are being attached to the cleaved DNA fragments and requires fewer library preparation steps than CUT&RUN. No additional annealing is necessary. The method works particularly well for nucleosomal and tightly bound proteins. It has also been streamlined by the Henikoff lab into a protocol where the entire process takes place in one tube and high throughput variations amenable for automation are available.
CUT&RUN on the other hand is preferable for transcription factors and other less tightly bound DNA binding proteins that are sensitive to the higher salt concentration in CUT&Tag necessary to prevent off-target tagmentation of accessible chromatin by Tn5. In addition, the spatial resolution of the MNase digestion is higher than that of the tagmentation, enabling a clearer footprint of the protein of interest.
Can CUT&RUN and CUT&Tag replace ChIP-seq? Does CUT&RUN work for transcription factors, do-factors and other indirect binders?
CUT&RUN and CUT&Tag work very reliably for abundant chromatin-associated proteins such as histones. Less abundant and less tightly bound antigens like e.g. some transcription factors or non-direct DNA binders that are part of larger complexes, can dissociate from DNA since they are not cross-linked. This is even more relevant for CUT&Tag due to the high salt concentration in the wash buffer necessary to avoid premature tagmentation. In addition, the overall signal of CUT&RUN and CUT&Tag is lower than for ChIP-seq. Generally, this is more than compensated by the superior signal-to-noise ratio when compared to ChIP-seq sequencing reads. However, for sparse targets this can be an issue.
Several variations of CUT&RUN and CUT&Tag address this potential issue depending on the target protein in question to widen the range of target proteins. (i) Light fixation can prevent the dissociation of transcription factors while assuring epitope accessibility. (ii) Isolation of nuclei can lead to signal amplification by preventing the antibody to bind to non-DNA-associated antigen in the cytoplasm. (iii) A secondary antibody can increase the local concentration of Fc fragments for binding of pA/G-MNase or pA/G-Tn5, thus amplifying the signal. It also extends the reach of the fusion enzyme when targeting larger chromatin binding complexes. (iv) Addition of a chaotropic agent to the elution buffer prevents retention of chromatin fragments in larger protein complexes.
CUT&RUN LoV-U (Low Volume – Urea) incorporates the nuclear extraction, the use of a secondary antibody, and urea in the elution buffer and has been shown to reliably detect interaction sites of the non-direct chromatin binder beta-catenin. You can find a detailed CUT&RUN LoV-U step-by-step protocol in our CUT&RUN and CUT&Tag Handbook.
Why is the DNA yield so low?
CUT&RUN and CUT&Tag are performed using low cell numbers and the background signal is considerably lower than e.g. for ChIP. This can make reliable measurements of the DNA concentration using a fluorometric assay or by capillary electrophoresis challenging.
To assess the success of the CUT&RUN and CUT&Tag methods it is recommended to include a reaction using an antibody against and abundant histone modification such as h4K27me ( ABIN6923144) or h4K4me3 ( ABIN2668472) as a positive control. DNA fragments prepared using such an antibody can be measured by capillary electrophoresis on a Bioanalyzer or Tapestation.
Fluorometric determination of the DNA concentration on a Qubit or Nanodrop fluorometer can be misleading because the method does not distinguish between the CUT&RUN or CUT&Tag DNA fragments and larger genomic DNA.
How do I choose a primary antibody for CUT&RUN or CUT&Tag?
Antibodies that are recommended for ChIP-seq do not necessarily work in CUT&RUN in CUT&Tag. In contrast to ChIP-seq, the antigen is generally in its native state without additional fixation. Unless an antibody has already been tested for CUT&RUN/Tag, a recommendation for a method in which the antigen is expected to be in a native state is helpful, e.g. immunofluorescence. Unless indicated otherwise, the recommended dilution for immunofluorescence is also a good starting point for the antibody’s concentration in CUT&RUN/Tag.
How can I validate that my primary antibody is working for CUT&RUN?
For a CUT&RUN experiment the validation data could include e.g. a Tapestation or Bionalyzer plot showing the size distribution and qPCR data showing target enrichment.
The DNA yield of a CUT&RUN experiment appears typically very low compared to ChIP-seq because of the lower initial sample size and the substantially lower DNA background. In particular for less abundant target protein the concentration is often times too low to be reliably measured using a fluorometric assay or by capillary electrophoresis. PCR amplification of small CUT&RUN products, i.e. less than 50 bp, can be problematic and is therefore not an option.
Once a sequencing library has been generated and sequenced map sequencing reads and verify the accumulation of reads at known binding sites.
Why do I need a negative control antibody? Why not just use a no-antibody control?
The MNase used for CUT&RUN is an endo- and exonuclease that will unspecifically bind and cleave unprotected DNA in hyper-accessible DNA, e.g. in regions surrounding regulatory elements. Free MNase will preferentially cut DNA within these hyper-accessible regions, thus potentially causing false positives and increase the background signal in general.
To avoid this undesired effect of untethered MNase, chromatin is randomly coated with the CUT&RUN guinea pig anti-rabbit IgG negative control antibody ( ABIN101961) prior to the addition of pAG-MNase ( ABIN6950951) to the samples. pAG-MNase is then tethered via its Protein A or Protein G portion to the antibody’s Fc fragment and background DNA fragmentation is dictated by the random antibody binding as opposed to the nuclease digestion of hyper-accessible DNA regions.
In CUT&Tag, omitting the primary antibody and only using the secondary antibody ABIN101961 for the no primary negative control has the same effect.
Can I replace the antibody negative control for CUT&RUN using a knock-out (or knock-down) of my protein?
Both controls are useful but address different aspects of the experiment and are therefore not interchangeable.
The CUT&RUN guinea pig anti-rabbit IgG antibody ( ABIN101961) is used as a negative control to establish a reference background for peak calling. This is necessary because of the sparse background signal in CUT&RUN samples compared to ChIP-seq samples. The ko (or kd) control on the other hand gives an impression of unspecific binding of the antibody directed against the protein of interest to other proteins. It is useful to avoid identification of false positive signals.
Do I need to use a secondary antibody for CUT&RUN and CUT&Tag?
Depending on the host species and isotype of the antibody and the Protein A and/or Protein G MNase fusion protein, a secondary antibody may be necessary for MNase binding.
Protein A has good high affinity to all rabbit IgG antibodies but low affinity to rat, goat and sheep IgG isotype antibodies and certain mouse IgG antibody subclasses, in particular IgG1. Protein G on the other hand binds well to the Fc region of mouse, goat, sheep, and most rat IgG. Its affinity to rabbit IgG however is lower than that of Protein A. When using pAG-MNase introduced with the improved CUT&RUN protocol it is therefore generally not necessary to use a secondary antibody. Use of the pA-MNase of the original protocol however might require the use of a secondary antibody raised in rabbit to assure efficient binding of the fusion protein to the antibody. When using MNase fused to both protein A and protein G it is however not necessary to use a secondary antibody to assure antibody binding.
Nonetheless, it can be advantageous to use a secondary antibody in CUT&RUN in context with larger chromatin binding protein complexes which might sterically interfere with the MNase accessing DNA. The secondary antibody extends the reach of the fusion enzyme when targeting larger chromatin binding complexes.
For CUT&Tag a secondary antibody is recommended to increase the local concentration of Fc fragment binding site in the vicinity of the intended transposition site around the antigen of interest. This step is necessary to increase the specific signal.
One of my antibodies is mouse. Has your pAG-MNase good affinity for mouse antibodies, or do you advise to use a rabbit anti-mouse secondary antibody?
The pAG-MNase will work well with your murine antibody. The addition of protein G to the MNase is primarily to accomodate the use of mouse IgG1 monoclonal antibodies that bind poorly to protein A. The other IgG isotypes bind well to either protein A or protein G.
Should I include heterologous spike-in DNA for quantitation?
The pAG-MNase ( ABIN6950951) is highly purified to not contain any contaminating DNA from the E. coli expression host. Therefore, it is recommended to use heterologous spike-in DNA as for quantitation and to compare results across different experiments.
When using a pA/G-MNase or pA/G-Tn5 that has been expressed in E. coli and purified in-house to a lesser degree it is generally not necessary to use a spike-in control. In this case, a sufficient amount of bacterial DNA is carried over to serve as control.
Is it possible to fix the cells prior to immobilization?
It is possible to fix samples for CUT&RUN and CUT&Tag, e.g. to avoid dissociation of less abundant transcription factor or larger protein complex from the DNA during the course for the experiment.
Fixation can be done with formaldehyde at a lower concentration of 0.1%. Cross-linking at 1% formaldehyde can actually reduce signal, possibly due to epitope masking. Other cross-linkers like DSP can also be used.
Are there protocols to make this work for tissue, where I cannot necessarily isolate single cells?
The MNase used for CUT&RUN also accepts RNA as substrate so it might be possible to adapt the CUT&RUN protocol for use on RNA binding proteins.
CUT&RUN and CUT&Tag are not necessarily restricted to samples containing single cells. However, tissues fragments must be sufficiently small to allow permeabilization of the cell membrane; e.g. Spatial-CUT&Tag works with fixed tissue sections.
Instead of the proteinase K digestion can I denature the proteins in the CUT&RUN product complexes by heat?
We recommend against this option: the DNA of interest is at his point present in a complex consisting of the DNA, the antigen, the corresponding antibody, and the pAG-MNase. Boiling this complex will likely precipitate the DNA together with denatured protein. This will also primarily affect the short CUT&RUN products and not the larger DNA molecules, leading to a decreased signal to noise ratio in your library and potentially also reducing the library’s complexity. This effect is further exacerbated because of the lower melting temperature of these short molecules compared to the longer contaminating DNA molecules.
What is preferable for DNA extractions prior to library preparation: extraction using phenol-chloroform or affinity purification using a column?
A potential issue when using SPRI beads for the DNA fragment clean-up is the carry-over of active Proteinase K, which can interfere with the downstream PCR amplification. Therefore, a phenol-chloroform extraction is preferable to assure complete denaturation of Proteinase K.
Alternatively, it is possible to include a chaotropic agent to the elution buffer to release chromatin fragments in situ, followed by a DNA purification on beads. Our CUT&RUN and CUT&Tag Handbook includes a detailed step-by-step protocol of CUT&RUN LoV-U using this approach.
Is it possible to do single-end instead of paired-end sequencing of the CUT&RUN libraries?
Single-end sequencing instead of paired-end sequencing is possible. However, it has drawbacks compared to paired-end sequencing: (i) For abundant targets like histone marks or transcription factors a large number of binding sites is expected. Paired-end sequencing facilitates unambiguous mapping to the correct genomic position. This additional information reduces the necessary sequencing depth. (ii) MNase will digest the target DNA until the section covered by the protein of interest. Paired-end sequencing will reveal this footprint while the information is lost in single-end sequencing.
CUT&RUN and CUT&Tag Webinars

Method Comparison: CUT&RUN, CUT&Tag and ChIP-seq
Presented by: Stefan Pellenz, Ph.D. (Product Manager, antibodies-online)

Reconstructing Wnt Signaling using CUT&RUN
Presented by: Claudio Cantù Ph.D. (Senior Lecturer, Docent, Department of Biomedical and Clinical Sciences, Division of Molecular Medicine and Virology, Linköping University
Hosted by: Stefan Pellenz, Ph.D. (Product Manager, antibodies-online)

Antibody Validation for CUT&RUN and CUT&Tag
Presented by: Stefan Pellenz, Ph.D. (Product Manager, antibodies-online)

CUT&RUN LoV-U uncovers tissue-specific genomic targets
Presented by: Pierfrancesco Pagella (Senior Postdoctoral Researcher),
Gianluca Zambanini (University Research Assistent)
Linköping University
Hosted by: Stefan Pellenz, Ph.D. (Product Manager, antibodies-online)
Download Additional CUT&RUN Resources
- Download Handbook: Method Overview, Protocols and Reagents for CUT&RUN and CUT&Tag
- Download Poster: Epigenome Profiling with CUT&RUN and CUT&Tag
- Download Flyer: Discover our CUT&RUN Sets
Additional CUT&RUN Resources
References
- : "An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites." in: eLife, Vol. 6, (2018) (PubMed).
- : "Reproducibility: Standardize antibodies used in research." in: Nature, Vol. 518, Issue 7537, pp. 27-9, (2015) (PubMed).
- : "RSC-Associated Subnucleosomes Define MNase-Sensitive Promoters in Yeast." in: Molecular cell, Vol. 73, Issue 2, pp. 238-249.e3, (2019) (PubMed).
- : "High-Resolution Chromatin Profiling Using CUT&RUN." in: Current protocols in molecular biology, Vol. 126, Issue 1, pp. e85, (2019) (PubMed).
- : "Profiling of Pluripotency Factors in Single Cells and Early Embryos." in: Cell, Vol. 177, Issue 5, pp. 1319-1329.e11, (2020) (PubMed).
- : "CUT&Tag for efficient epigenomic profiling of small samples and single cells." in: Nature communications, Vol. 10, Issue 1, pp. 1930, (2019) (PubMed).
- : "Improved CUT&RUN chromatin profiling tools." in: eLife, Vol. 8, (2019) (PubMed).
- : "XL-DNase-seq: improved footprinting of dynamic transcription factors." in: Epigenetics & chromatin, Vol. 12, Issue 1, pp. 30, (2020) (PubMed).
- : "Efficient low-cost chromatin profiling with CUT&Tag." in: Nature protocols, (2020) (PubMed).
- : "Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation." in: eLife, Vol. 9, (2020) (PubMed).
- : "Spatial-CUT&Tag: Spatially resolved chromatin modification profiling at the cellular level." in: Science (New York, N.Y.), Vol. 375, Issue 6581, pp. 681-686, (2022) (PubMed).
- : "An improved CUT&RUN method for regulation network reconstruction of low abundance transcription factor." in: Cellular signalling, Vol. 96, pp. 110361, (2022) (PubMed).
- : "A new cut&run low volume-urea (LoV-U) protocol optimized for transcriptional co-factors uncovers Wnt/b-catenin tissue-specific genomic targets." in: Development (Cambridge, England), (2022) (PubMed).

Goal-oriented, time line driven scientist, proficiently trained in different academic institutions in Germany, France and the USA. Experienced in the life sciences e-commerce environment with a focus on product development and customer relation management.
Go to author page


